Code
Code Availability
Most code below is available at https://gitlab.com/barker-lab unless otherwise indicated below.
EvoPipes
A repository of the core EvoPipes pipelines is available at https://gitlab.com/barker-lab/EvoPipes. This is a suite of bioinformatic tools for whole genome duplication inference, HMM-based gene translation, and ortholog identification. Originally hosted at evopipes.net, this repository provides all code and a Docker that provides a fully functioning distribution of EvoPipes. It is recommended that most users run the Docker version.

Frackify
Machine learning classification of gene duplication origins using gradient boosted decision trees and XGBoost. Combines synteny and divergence features to identify paleologs in diploidized species with complex duplication histories, enabling users to classify gene duplications as paleologs with different levels of retention or as other duplication types. Available through GitLab (https://gitlab.com/barker-lab/frackify) and Docker (https://hub.docker.com/r/mmckibben/frackify). Publication: McKibben & Barker 2023
Ploidify
Logistic regression model for identifying ploidal levels of ancient whole genome duplications using Frackify output data. Analyzes paleolog retention patterns to classify ancient WGDs as tetraploid versus hexaploid events, providing insight into the nature of ancient genome duplication events across evolutionary time. Kang et al. in prep.
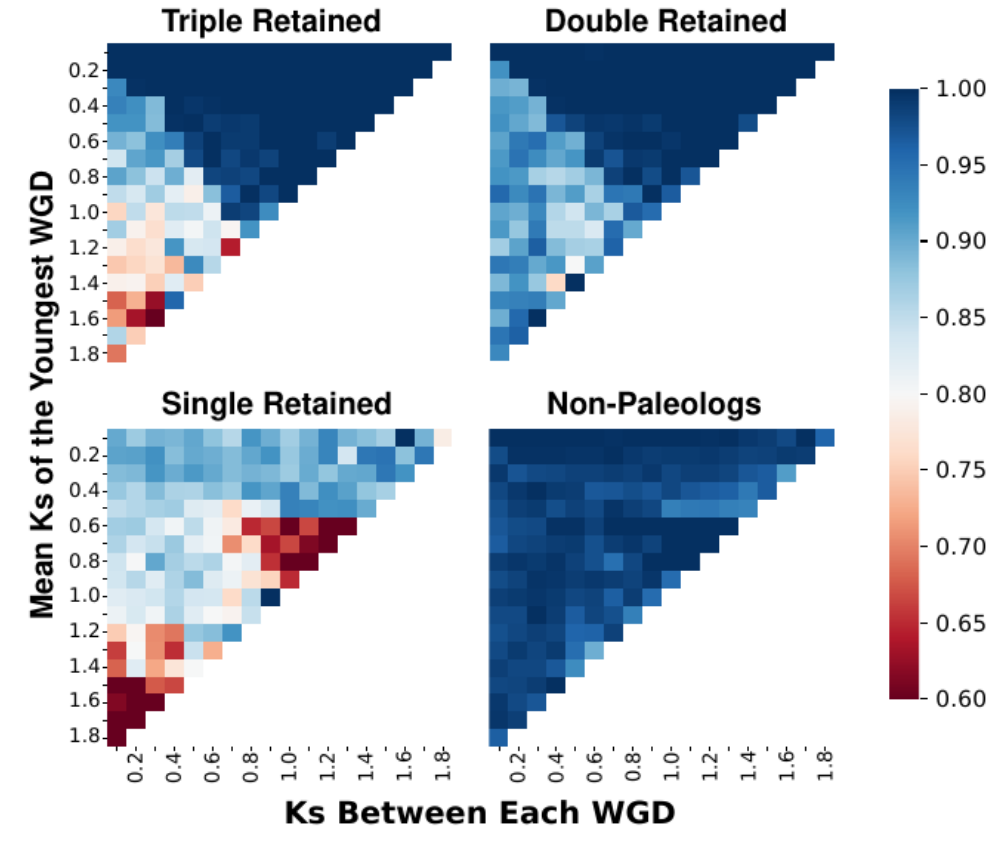
Heat maps of Frackify’s classification accuracy on single, double, and triple retained genes in simulations containing three independent WGDs of varying divergences from each other. After McKibben and Barker 2023.
SLEDGE
Machine learning tool for automated detection and classification of ancient whole genome duplications in paralog Ks distributions. Uses a variety of algorithms to identify WGD signals from phylogenomic data, enabling large-scale analysis of genome duplication events across diverse taxonomic groups. Publication: Sutherland et al. 2024 bioRxiv

Sample of simulated WGDs used for training machine learning models of WGD inference in forthcoming tool SLEDGE (Sutherland et al. in prep).
HyDe-CNN
Convolutional neural network approaches for classifying hybridization events and hybrid speciation patterns in genomic data. Uses coalescent simulations to train networks that perform model selection for hybridization scenarios using matrices of pairwise nucleotide divergence calculated across genome windows, enabling automated detection and classification of complex hybridization signatures in large-scale genomic datasets. Available through GitHub (https://github.com/pblischak/hyde-cnn). Publication: Blischak et al. 2021, Molecular Ecology Resources
dadi Models for Polyploids
Extended demographic inference models for polyploid and inbred organisms, developed by Paul Blischak. Uses diffusion approximation methods adapted for complex mating systems, providing formal approaches for estimating degrees of tetrasomic versus disomic inheritance and enabling population history reconstruction in crops and polyploid species. Available through GitHub repositories: inbreeding models (https://github.com/pblischak/inbreeding-sfs) and polyploid models (https://github.com/pblischak/polyploid-demography). Publications: Blischak et al. 2020, Molecular Biology and Evolution; Blischak et al. 2023, Genetics
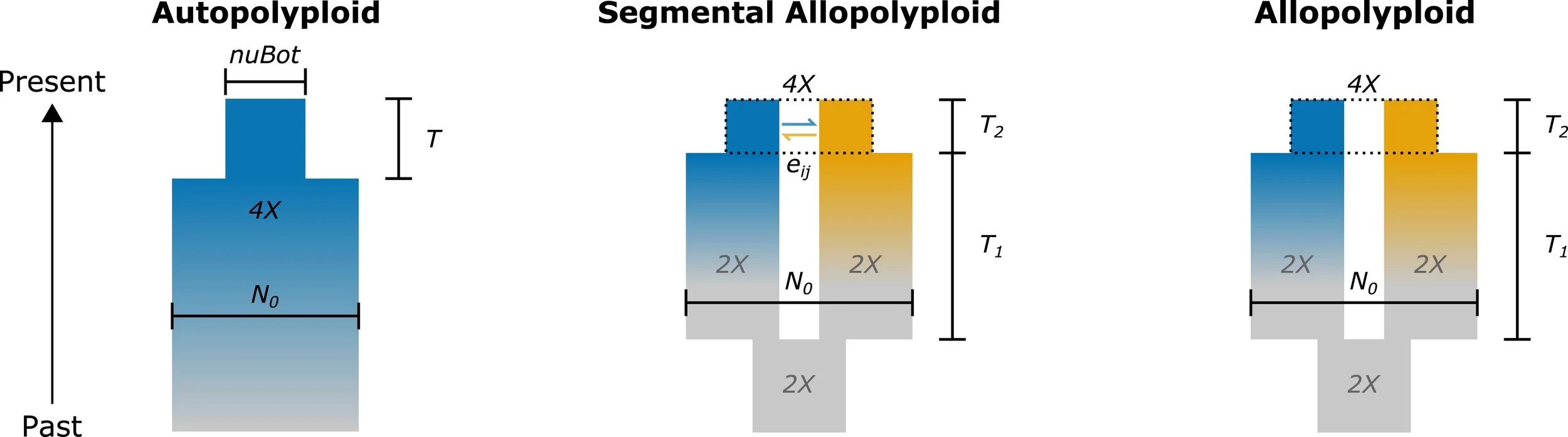
Graphical representations of the models used in the Blischak et al. 2023 dadi models for validating the diffusion approximation for autopolyploids (left), segmental allopolyploids (middle), and allopolyploids (right).
Animal Chromosome Count Database
The Animal Chromosome Count Database (ACC) is the largest database of animal chromosomal counts. We have curated chromosome numbers across the animal Tree of Life to make data accessible for people interested in understanding the potential links between biological processes and patterns to broad chromosomal changes in animals. Available through GitHub (https://github.com/cromanpa94/ACC) and web interface (https://cromanpa94.github.io/ACC/). Publication: Román-Palacios et al. 2021, Journal of Evolutionary Biology

Distribution of animal chromosome counts in the ACC.
GOgetter
Functional annotation tool for analyzing gene ontology patterns in plant genomes using hierarchical Gene Ontology (GO) Resource annotations. Provides fast and easy-to-implement pipeline for obtaining, summarizing, and visualizing GO slim categories associated with gene sets, enabling comprehensive analysis of functional consequences of genome duplications and gene retention patterns. Available through GitHub (https://github.com/jessiepelosi/GOGetter). Publication: Sessa et al. 2023, Applications in Plant Sciences
Tools in Development
SynTRACE
Synteny Tracking Relationships Across Collinear Evolution - a GPU-accelerated tool for large-scale syntenic network analysis. Uses community detection algorithms to identify conserved genomic building blocks across hundreds of species simultaneously. Enables quantitative analysis of genome structural evolution rates and identification of syntenic blocks conserved across phylogenetic scales, providing new insights into the fundamental organization principles of plant genomes.
GOmosaic
Enhanced multi-dataset GO term visualization tool with LCA-based cluster naming and accessibility-optimized color management. Provides coordinated visualization across multiple experimental conditions using treemap layouts with consistent color families for semantic clusters. Features automatic biological theme identification through Lowest Common Ancestor methodology and enhanced GOGO semantic similarity clustering. Enables comparative functional genomics analysis while maintaining inclusive design.
Legacy Tools
MAPS: Multi-tAxon Paleopolyploid Search algorithm
Phylogenetic placement algorithm for ancient whole genome duplications. Provided statistical methods for placing WGDs on phylogenies, now largely superseded by newer approaches for WGD inference and classification. Publications: Li et al. 2015, Science Advances; Li et al. 2018, PNAS

Flow chart of the MAPS algorithm (Li et al. 2015) from Li et al. in preparation.
NU-IN
Tool for simulating gene family evolution with real sequence data. Enabled modeling of gene duplication and loss dynamics within a phylogenetic framework using empirical sequence information to inform evolutionary simulations. Publication: Barker 2010, BMC Research Notes
SCARF
Transcriptome assembly tool developed for improving RNA-seq assembly quality and accuracy during the early phases of transcriptomic analysis. Historical tool that contributed to enhanced transcriptome assembly methods before current-generation assembly algorithms became standard. Publication: Barker et al. 2009, Bioinformatics